21. Clasificación Incompleta del Linaje y Tamaños de Población Ancestrales
1 de Agosto de 2013. Temas: Genética
Nota: Esta serie de artículos ha sido concebida como una introducción básica a la ciencia de la evolución para no especialistas. Aquí se puede ver la introducción a esta serie o volver al índice aquí.
En este artículo discutimos cómo pueden utilizarse los datos de la genómica comparada para estimar los tamaños poblacionales en diferentes puntos dentro de una filogenia.
En el último artículo de esta serie introducíamos el difícil (desafiante) concepto de los árboles de genes discordantes con los árboles filogenéticos de especies, que surgen por clasificación del linaje incompleta1 (ILS). En este artículo, vamos a echar un vistazo a una de las implicaciones interesantes del ILS, su utilización para estimar tamaños de población, antes de pasar a otros temas relacionados. (De nuevo, si este tema parece demasiado espinoso, podemos saltar este artículo y el siguiente, porque los artículos posteriores de la serie no dependen de la comprensión de esta información).
Podemos utilizar la ILS para calcular tamaños de población porque los árboles discordantes nos proporcionan la forma de medir el número de alelos presentes en la población ancestral que, a su vez, pueden utilizarse para estimar el número de individuos de la población. Sin embargo, antes de entrar en detalles, revisemos brevemente cómo la especiación es un fenómeno de nivel poblacional.
Como recordaremos de anteriores artículos de esta serie, los procesos de especiación comienzan cuando dos poblaciones quedan genéticamente aisladas entre sí, bien completamente o bien sólo en parte. Esto permite que las características medias de las dos poblaciones diverjan, lo que con el tiempo conduce a la especiación. Lo que hay que enfatizar aquí es el hecho de que las dos poblaciones son poblaciones: un grupo de organismos de la misma especie que se reproducen entre sí. Las poblaciones, como hemos visto, son capaces de transmitir mucha más diversidad genética que un solo organismo individual; un individuo sólo puede tener dos alelos de un determinado gen, mientras que una población puede tener centenares, o incluso miles.
Árboles discordantes: una ventana al pasado
Con esto en mente podemos volver a nuestra discusión sobre la clasificación del linaje incompleta y sobre los árboles resultantes de genes discordantes que se encuentran dentro de un árbol de especies. El ejemplo que utilizábamos anteriormente presentaba al gorila y al chimpancé con alelos más cercanamente relacionados, y al hombre con alelos más lejanamente relacionados:
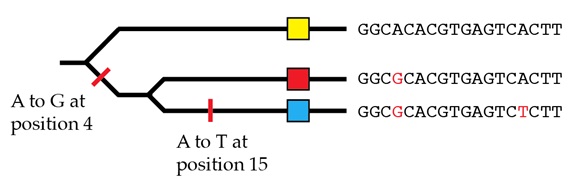
Luego describíamos un ejemplo de clasificación del linaje incompleta en el que gorilas y chimpancés heredan los alelos más cercanamente relacionados, y el hombre hereda un alelo más lejanamente relacionado:
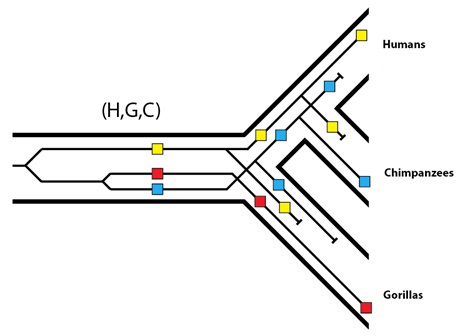
Ahora podemos discutir qué es lo que podemos deducir de este patrón, y qué nos dice sobre la población ancestral común (H, G, C). Lo primero que nos dice este patrón es que los alelos rojo y azul estaban presentes antes de que el linaje del chimpancé se separara del linaje del gorila. Como sabemos por el árbol de especies que la población ancestral común gorila / chimpancé es la población ancestral común (H, G, C), esto confirma que los alelos rojo y azul eran parte de la variación que esta población mantenía. A continuación, hay que subrayar que el alelo amarillo es más ancestral; en otras palabras, tiene menos mutaciones en comparación con los alelos rojo y azul. Ello significa que el alelo amarillo es más antiguo que los alelos rojo y azul. Esto sitúa al alelo amarillo, en la filogenia, con anterioridad al proceso de especiación (G) / (H, C). Además, como los humanos tenemos el alelo amarillo, tiene que haber estado presente en la población ancestral común (H, C) en el momento de su separación del linaje (G). Considerando todo ello en conjunto, vemos que el alelo amarillo estaba también presente en la población (H, G, C). En ausencia de nuevas mutaciones, que han sido excluidas en estos análisis, no hay ninguna otra forma de producir este patrón de herencia a menos que los tres alelos estuvieran presentes en la población (H, G, C). Aunque las tres especies en la actualidad tengan cada una sólo uno de los tres alelos, podemos inferir que su población ancestral común tenía los tres.
Así que, los árboles de genes discordantes son una ventana abierta al pasado que revela la diversidad genética de una población ancestral; cuántos alelos mantenía en una determinada región del genoma. Comparando grandes muestras de datos de genoma de humanos, chimpancés y gorilas, es posible obtener una estimación precisa del tamaño poblacional de la población ancestral (H, G, C) (alrededor de unos 50.000 individuos). Esta medida, conocida como tamaño de población efectivo (simbolizado Ne) es el tamaño de población necesario para transmitir la cantidad de variación genética observada desde una población ancestral hasta nuestros días. El linaje ancestral común humano / chimpancé (H, C), estimado con los mismos métodos, también da alrededor de unos 50.000 individuos a lo largo de su historia.
Comprobación del modelo con una nueva especie adicional: el genoma del orangután
La secuenciación del genoma del orangután, terminada en 2011, proporcionó a los investigadores la oportunidad de comprobar estas estimaciones utilizando un nuevo conjunto de datos. El linaje del orangután se ramifica, en la filogenia de los primates, de una población ancestral común (H, O, G, C; en la que la “O” hace referencia al orangután) abandonando la población ancestral (H, G, C) que más tarde sufriría la especiación:
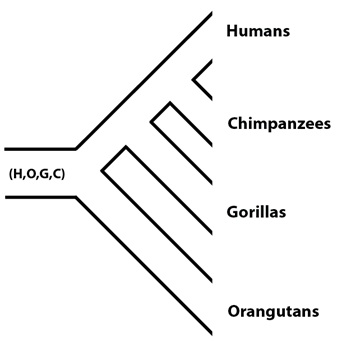
Utilizando las estimaciones precedentes de tamaños poblacionales de (H, G, C) y de (H, C), los investigadores fueron capaces de predecir de antemano que una parte muy pequeña de los genomas humano y del orangután deberían estar más estrechamente relacionados entre sí; es decir, que la clasificación del linaje incompleta debería haber producido algunas raras regiones del genoma en las que los alelos del hombre y del orangután fueran más parecidos entre sí que con los de otros primates. El valor esperado de esos pares de regiones (H, O), de aproximadamente un 1,2%, es muy pequeño si se compara con el valor predicho para los pares de regiones (H, G), que es de alrededor de un 25%, en gran parte porque los humanos, los chimpancés y los gorilas sufrieron la especiación en un lapso de tiempo relativamente corto, mientras que el tiempo que pasó desde la divergencia del orangután hasta la posterior divergencia del gorila fue comparativamente mayor. La fracción de nuestro genoma que más se parece a otra del genoma del orangután es de alrededor de un 0,8%; valor notablemente cercano al predicho y coherente con los valores de Ne estimados para las poblaciones (H, G, C) y (H, C) del trabajo anterior.
En otras palabras: al comparar los genomas de los primates, vemos un patrón de clasificación del linaje incompleta. Tal como esperábamos, nuestro genoma coincide mucho con el del chimpancé, algo menos con el del gorila, y todavía menos con el del orangután. Por otra parte es formalmente posible que, cuando algún día se secuencie y se analice el genoma del gibón, pueda haber trazas de clasificación del linaje incompleta que produzcan agrupaciones de alelos (humano, gibón); aunque es probable que esta fracción del genoma resulte demasiado pequeña como para ser detectada de forma fiable, porque los gibones se ramificaron del árbol de los primates mucho antes todavía que los orangutanes.
Resumiendo y mirando al futuro
Lejos de resultar un “problema” para el modelo o patrón del antecesor común, la clasificación del linaje incompleta es una consecuencia esperada de las poblaciones sometidas a procesos de especiación y una ventana a su diversidad genética. El resultado final en una filogenia es, como hemos visto, un subconjunto de características que forman un árbol discordante dentro del árbol filogenético de especies. En el artículo siguiente de esta serie exploraremos otro efecto que puede también producir patrones que no concuerden con el árbol filogenético de especies: la evolución convergente.
Lecturas complementarias
Hobolth A, et al., (2007). Genomic Relationships and Speciation Times of Human, Chimpanzee, and Gorilla Inferred from a Coalescent Hidden Markov Model. PLoS Genet 3(2): e7 (fuente)
Holboth A., et al. (2011). Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection. Genome Research. 2011 March; 21(3) 349. (fuente)
Notas
- Nota del T.: En el original “ILS= incomplete lineage sorting”


