19. Los Genomas como Textos Antiguos, Parte 5
18 de Julio de 2013. Temas: Genética
Nota: Esta serie de artículos ha sido concebida como una introducción básica a la ciencia de la evolución para no especialistas. Aquí se puede ver la introducción a esta serie o volver al índice aquí.
En este artículo se revisan las diversas líneas de evidencia de un antecesor común procedentes de la genómica comparada, y vemos que son coherentes con un modelo o patrón mutuamente cohesionado.
Los genomas como textos copiados: relacionarlo todo
A lo largo de los últimos artículos de esta serie hemos venido examinando los patrones generales que observamos al comparar los genomas de distintas especies entre sí. Lo que hemos visto es que el patrón que observamos es del todo coherente con el hecho de que las especies compartan antecesores comunes- y sus genomas, de acuerdo con ello, sean copias modificadas de lo que una vez fue el genoma de la población antecesora común. Lo que todavía no hemos discutido en detalle, hasta ahora, es que las evidencias que hemos examinado, comparaciones de la estructura del genoma, secuencias génicas funcionales, y mutaciones específicas en genes inactivados, todas son coherentes en un patrón en el que se apoyan unas a otras. La cohesión de múltiples e independientes evidencias, es precisamente en lo que está basada una buena teoría, y la Genómica comparada ha proporcionado un apoyo realmente sólido a la teoría evolutiva.
Empezando por los “errores tipográficos” compartidos
Volvamos un momento sobre el ejemplo de los genes desactivados de los receptores olfativos que discutíamos en nuestro artículo anterior. Basándonos sólo en ese pequeño grupo de genes defectuosos, hemos construido el siguiente “árbol genealógico” de la especie humana, de los chimpancés, de los gorilas y de los orangutanes:
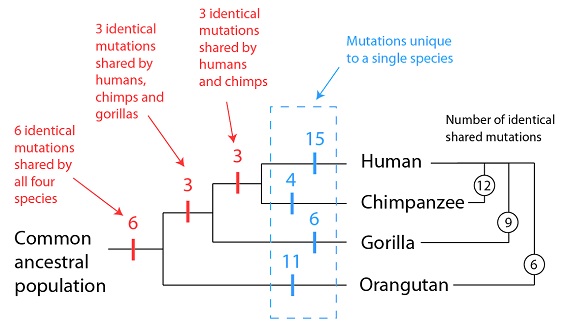
Un árbol genealógico que se propone para relacionar varias especies,-- o árbol filogenético, por introducir el término científico--, es una forma gráfica de (a) representar un gran conjunto de datos, y (b) proponer una hipótesis de cómo ese conjunto de datos llegó a producirse. En ese conjunto de datos, tenemos que explicar dos categorías de caracteres: las mutaciones que son idénticas en más de una especie, y las mutaciones que son únicas en una sola especie. Y como ya hemos tratado antes, la filogenia anterior encaja muy bien con los datos; los patrones de los sucesos compartidos y de los sucesos únicos quedan apoyados por la misma filogenia. Los sucesos compartidos tienen lugar una única vez, en un genoma común y los sucesos únicos se dan después de que dos especies hayan tomado ya caminos separados.
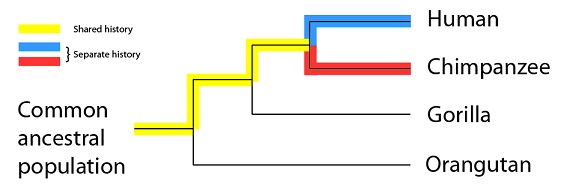
Lo que en realidad está proponiendo esta filogenia es que estas cuatro especies tienen distintas duraciones de historia compartida, y también de historia separada. Por ejemplo, los humanos y los chimpancés tendrían el máximo de tiempo de historia compartida de estas cuatro especies (subrayado en amarillo), y un tiempo comparativamente corto de historias separadas (subrayadas en azul y en rojo):
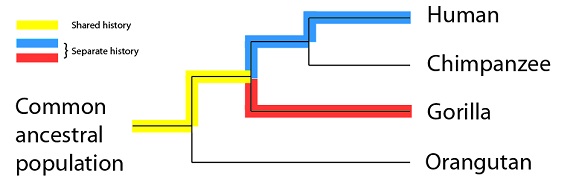
Humanos y gorilas, sin embargo, tienen menos tiempo de historia compartida, y más de historia separada en el mismo lapso de tiempo:
Y finalmente, orangutanes y humanos tienen todavía menos tiempo de historia compartida con los otros primates, y más de historia separada:
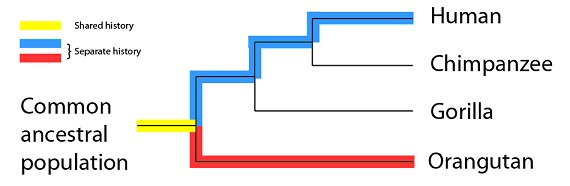
De manera que, con sólo esta pequeña muestra, un puñado de mutaciones compartidas en unos pocos genes, tenemos una hipótesis detallada sobre qué especies comparten más tiempo de historia en común; una hipótesis que también podemos comprobar con otro tipo de evidencias.
De los errores tipográficos a las frases
Ahora que ya hemos manejado una pequeña submuestra de los “errores tipográficos compartidos” encontrados en estos cuatro genomas para elaborar una propuesta de filogenia, podemos considerar qué es lo que esta filogenia predeciría al comparar las secuencias de genes individuales en estas cuatro especies. La clave es la parte de historia compartida entre dos especies en la filogenia: durante ese tiempo, lo que luego van a ser dos especies, es todavía una única población, con un genoma común. Por lo tanto, ambas tendrán la misma secuencia para un gen cualquiera, a no ser que haya variación para ese gen en la población, en cuyo caso la población compartirá un grupo de alelos de ese gen. Cuanto más largo sea el tiempo de historia compartida entre las dos especies, más parecida podemos esperar que sea la secuencia de genes. Y cuanto más tiempo hayan tenido de historia separada, tanto mayor será la diferencia que podemos esperar entre sus genes, debido a las mutaciones que tienen lugar durante la parte de “historia separada” de su filogenia.
Al secuenciar el genoma del orangután en 2011 y el genoma del gorila en 2012, podemos ya comprobar esta predicción utilizando un gran número de datos de las cuatro especies. Las secuencias humana y del chimpancé son, en general, casi idénticas (98.6% de identidad); entre humanos y gorilas un poco menos (98,3% de identidad) y entre humanos y orangutanes aún menos (96.6% de identidad). Estos resultados encajan dentro del patrón o modelo predicho:
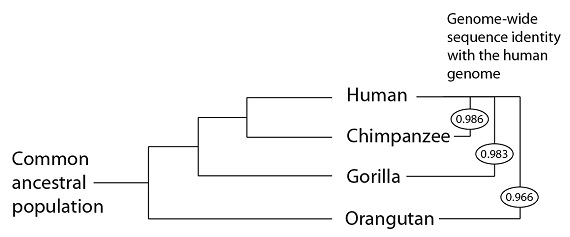
Identidad de secuencias de de los genomas respectivos el genoma humano Hombre
Población Chimpancé
ancestral Gorila
común Orangután
En otras palabras, la filogenia a la que se llega, para estas especies, utilizando grandes segmentos de secuencias de genoma es la misma filogenia que la que proporcionan los datos de las mutaciones compartidas. El patrón que requieren los datos de mutaciones compartidas, (una pequeña submuestra de secuencias del ADN de estas especies), es también el patrón que mejor explica la identidad global del genoma que observamos.
De las frases a los capítulos
Con los datos de mutaciones compartidas y los de secuencias globales apoyando la misma filogenia para estas especies, podemos ir más allá comprobando ahora estas hipótesis mediante la estructura del genoma (la organización espacial de los genes en los cromosomas, o los “capítulos”, volviendo a nuestra analogía del libro copiado). Como en el caso de los datos de mutaciones compartidas, las similitudes y las diferencias que observamos se espera que caigan o entre las características compartidas (en el modelo o patrón predicho) o bien que sean características únicas (que surgieran una vez que la especie se hubiera separado de otra especie). En conjunto, al comparar la estructura cromosómica de las cuatro especies1, comprobamos que todo eso se cumple. Al comparar la estructura cromosómica, los humanos a lo que más se parecen es a los chimpancés, algo menos a los gorilas, e incluso menos todavía a los orangutanes, tal como esperábamos. Para ilustrar este patrón con un ejemplo específico, volvamos a la mayor diferencia estructural cromosómica existente entre los humanos y los grandes simios, la fusión que dio lugar al cromosoma 2 humano. Como ya hemos tratado previamente, el cromosoma fusionado está presente en humanos, pero no en chimpancés, ni en gorilas, ni en orangutanes; lo cual quiere decir que tuvo lugar después de que el linaje humano se separara del linaje del chimpancé. Un examen más detallado de esta región en los gorilas y orangutanes demuestra que hay otra diferencia: una parte de dicha región está invertida en los genomas humano y de chimpancé (marcado en rojo en el esquema) cuando se comparan con la región equivalente en gorilas y orangutanes (marcado en azul en el esquema):
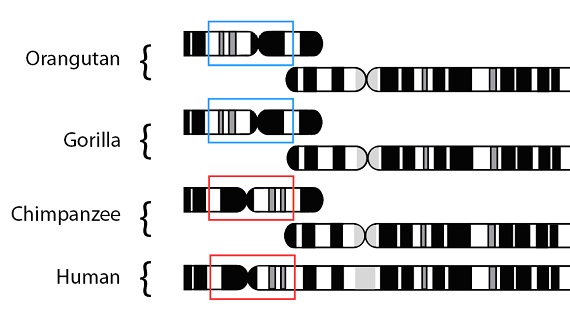
Así que, los humanos son los más parecidos a los chimpancés, (la mayoría de las regiones coinciden), y menos a las otras especies de monos, como se esperaba. Las diferencias que vemos se mapean fácilmente también en la filogenia construida por medio de otras evidencias. Como la inversión es común a humanos y chimpancés, pero no aparece en las otras especies, es probable que apareciera en la población ancestral común al hombre y al chimpancé, después de que ésta se separara del linaje que lleva a los gorilas. La fusión tendría lugar más tarde, en el linaje que conduce a la especie humana (y que, como hemos visto, es compartido por otras especies más estrechamente emparentadas con la especie humana que con los grandes simios). Como esperábamos, la filogenia construida utilizando sólo los datos de estructura cromosómica coincide con la filogenia predicha por otros tipos de evidencias:
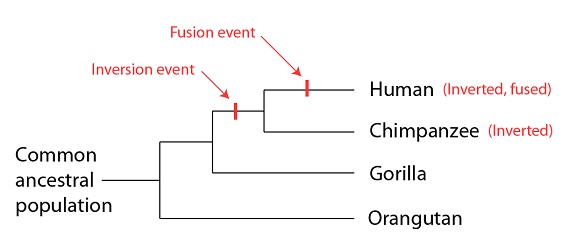
Fusión
Inversión
Hombre (Invertido, fusionado)
Población Chimpancé (Invertido)
ancestral Gorila
común Orangután
Resumiendo y mirando al futuro
Como discutíamos al empezar esta serie de artículos, una buena teoría, en sentido científico, es la que resulta apoyada por múltiples evidencias y realiza predicciones precisas. Con la aparición de la moderna genómica comparada, la teoría evolutiva ha mostrado ser sólida en una forma tal que Darwin no pudo ni siquiera imaginar. Podemos decir con confianza que compartimos antecesores con otras especies, y esta conclusión no es en absoluto probable que vaya a cambiar, ni siquiera cuando aparezca información nueva.
En el artículo siguiente de esta serie, vamos a dirigir nuestra atención sobre las características que no encajan demasiado bien en las filogenias predichas. Lejos de constituir un problema, como a menudo se alega por los detractores de la evolución, dichas características son un cúmulo de información que incluso revela aún más cosas sobre nuestro pasado.
Referencias citadas:
1 Diagrams of late prophase chromosomes (1000-band stage) of human, chimpanzee, gorilla, an orangután (left to right for each number). Courtesy of Jorge Yunis. En: Yunis, J.J. y O. Prakash. 1982. From “The Origin of Man: A Chromosomal Pictorial Legacy. In Science 215: 1525-1530.


